What is a Phylogeny Tree?
A phylogenetic tree is a representation of when species diverged from one another by connecting them by most recent common ancestry [1]. This most recent common ancestry can be inferred using Maximum Likelihood which uses evolutional probability of nodal (most recent ancestral) connection [2].
A phylogenetic tree can be made using MEGA11 software where Clustal Omega or MUSCLE are used to align sequences before the Maximum Likelihood test is completed.
A phylogenetic tree can be made using MEGA11 software where Clustal Omega or MUSCLE are used to align sequences before the Maximum Likelihood test is completed.
Phylogeny Trees of FOXN1

neighbor-joining tree aligned by clustalw
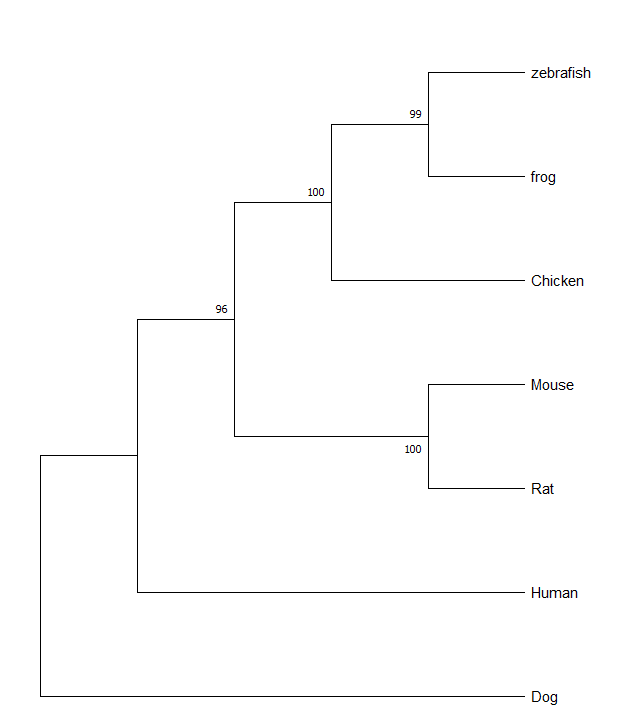
neighbor-joining tree aligned by MUSCLE
|

maximum likelihood tree aligned by clustalw

maximum likelihood tree aligned by MUSCLE
|
Discussion
The phylogenetic trees for FOXN1 reflect that human FOXN1 is most similar and most likely the most related to dog FOXN1 and is then most similar to mouse and rat FOXN1. While it shows that these models may have a more similar FOXN1 gene to humans, that does not necessarily mean that these are the best models for this research.
Sources
1. Nature Publishing Group. (n.d.). Reading a Phylogenetic Tree: The Meaning of Monophyletic Groups. Nature news. https://www.nature.com/scitable/topicpage/reading-a-phylogenetic-tree-the-meaning-of-41956/
2. Maximum likelihood method. Maximum Likelihood Method - an overview | ScienceDirect Topics. (n.d.). https://www.sciencedirect.com/topics/medicine-and-dentistry/maximum-likelihood-method#:~:text=Maximum%20likelihood%20methods%20(Felsenstein%2C%201981,which%20statistical%20significance%20is%20inferred.
IMAGES:
Biorender
2. Maximum likelihood method. Maximum Likelihood Method - an overview | ScienceDirect Topics. (n.d.). https://www.sciencedirect.com/topics/medicine-and-dentistry/maximum-likelihood-method#:~:text=Maximum%20likelihood%20methods%20(Felsenstein%2C%201981,which%20statistical%20significance%20is%20inferred.
IMAGES:
Biorender
This web page was produced as an assignment for Genetics 564, a capstone course at UW-Madison.
